
这部分内容还是第一次转录组的实操练习,之前已经完成了序列的拼接、去冗余,并且已经做了层次聚类,统计了每个transcript的raw counts,现在进入了差异基因分析的步骤。因为不管采用什么软件,本质的方法是一致的,因此也具有一定的通用性,或者可以迁移到其他基因分析的项目中,因此要用心学习。
前言
这里采用的是R中的edgeR包来进行分析,因此需要先做环境准备,现在开始。
1. 环境准备
- 安装R,可以到官网download
- 安装相应包,如下
if (!requireNamespace(“BiocManager”, quietly = TRUE))
install.packages(“BiocManager”)
if (!requireNamespace(“edgeR”, quietly = TRUE))
BiocManager::install(“edgeR”)
if (!requireNamespace(“airway”, quietly = TRUE))
BiocManager::install(“airway”)
2. 全过程代码
1 | # 导入依赖包 |
成果示例:
总结
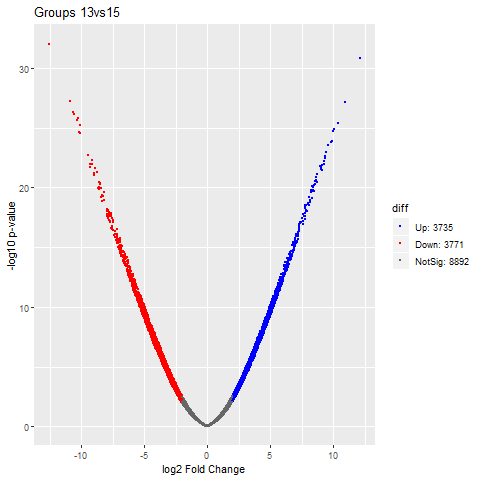
- 在这个包的分析过程中,可以调整dispersion/pvalue/logFC这3个参数以及CPM 标准化的过滤阈值来调控我们最终获取的差异基因的数量,一般我们能分别获得几千个上下调基因就可以了,可以适当调整参数,方便下一步分析。
- 在提取出来后,我们只是得到对应的id和它的down或者up标签,还需要把他们从原来的fasta文件中提取出来,这样才能接着做blast分析,这里使用的工具是TBtools这个软件,可以很好的做到这一点,但是建议还是得自己编程摸索一下,避免对别人的软件过渡依赖。
- 在Linux中还有专门的split命令,可以直接一句命令进行文件拆分,拆分的依据可以是行数,也可以是大小(如10M),还可以是其他标准,这个非常方便,能够快速的进行拆分,然后我们只需要对拆分的文件进行重命名就好了。
- 在使用TBtools的时候,要注意的是,原始的fasta文件不要把其中的id前面的“>”去掉了,去掉后,这个软件没有办法定位哪里是序列开头,这样子就会导致提取序列出错。