
继上一篇edgeR基因差异分析,通过采用R中的edgeR/limma这两个包,完成了差异基因的分析。在这个分析中总共分为了9组进行差异分析,最终结果取了并集,然后对并集进行blast,进而统一进行blast2go注释,再对每一组差异分析单独的从结果中匹配出相应的注释。现在,我需要做的就是这个并集的blast,为blast2go做准备。值得注意的是,这里的blast效率可能并不高,因此会耗费大量时间,可以考虑拆分文件,开启多个进程同时比对。
1. blast简介
blast全称是Basic Local Alignment Search Tool,是NCBI运营的一个基础序列比对软件,通过这个软件可以做到很好的多序列比对,比对到蛋白质或者核酸数据库中。众所周知,序列比对是生信分析的基础,不管是哪一个组学的内容,都必须进行比对才能开启下一步的工作。blast不是单纯的一个program,而是多个program的综合称呼:
|program|progress|
|:—-|:—-|
|blastn|nr–>nr|
|blastx|nr–>pro|
|tblastn|pro–>nr|
|blastp|pro–>pro|
现在在这个项目中,我们需要做的下一步是比对到nr数据库,nr数据库是一个蛋白质序列库,我们的序列是核酸序列,因此我们使用的是blastx program,即核酸比对到蛋白质。还有另外一个数据库——nt,这个数据库是一个核酸数据库,如果需要比对到核酸的话,就可以选择这个数据库进行比对。
2. blast使用
如果是第一次使用blast,那么需要做一些前期的准备工作,不过这个准备工作是通用的,完成一次后,下一次可以直接使用,现在我们从准备工作开始做起。
2.1 下载安装blast
可以进入官网寻找到下载地址ftp://ftp.ncbi.nlm.nih.gov/blast/executables/blast+/LATEST/,点击网址进入选择合适的版本,download到自己的服务器或者本地中。
- 解压blast文件,将bin目录下内容copy到/usr/local/bin中
这一步的目的是为了让blast能够直接在服务器任何一个地方使用,而不用输入绝对路径,相当于是添加到了环境变量中,因为/usr/local/bin就是环境变量中的一个,所以同样也可以通过添加新环境变量的方式来达到以上目的。
- 制作软链接,将解压后的的bin目录软链接到/home/username
- 编辑.bashrc文件,添加
export PATH=/home/username/bin/:$PATH;,即添加环境变量2.2 准备数据库
在这里除了blast程序之外,还得有本地的数据库才行。so,第一步,先下载blast对应的一些数据库。 - 下载数据库(nr nt等):ftp://ftp.ncbi.nlm.nih.gov/blast/
也可以先浏览一下这个网站:https://www.ncbi.nlm.nih.gov/books/NBK62345/
- 通过2.1中下载的blast软件构建索引|makeblastdb
makeblastdb -in mature.fa -input_type fasta -dbtype nucl -title miRBase -parse_seqids -out miRBase -logfile File_Name
| options | explain |
|---|---|
| -in | 你要格式化的fasta序列 |
| -dbtype | database的类型,nucl为核酸,prot为蛋白 |
| -title | 给数据库起名字,不能作为-db的参数使用 |
| -parse_seqids | 暂时未知 |
| -out | 给数据库起个名字,能在blast+时作为-db的参数使用 |
| -logfile | 日志文件,默认输出到屏幕 |
2.3 开始使用blast
最好保持一个良好习惯,先新建好文件目录,整理好文件,做好记录,然后再进行blast比对。
nohup blastx -query ../up1.fasta -db /B313/public_db/blast/nr -outfmt 5 -num_threads 20 -out up1-blastx >log1.out 2>&1 &
nohup blastx -query ../up4.fasta -db /B313/public_db/blast/fish_prot -outfmt 5 -num_threads 8 -out up4-blastx >log4.out 2>&1 &
nohup blastx -query ../down1.fasta -db /B313/public_db/blast/fish_prot -outfmt 5 -num_threads 8 -out down1-blastx >dlog1.out 2>&1 &
nohup blastx -query ../gene_data/53vsX-downgenes.fasta -db /B313/public_db/blast/fish_prot -outfmt 5 -num_threads 8 -out 53vsXdown-blastx >dlog_53.out 2>&1 &
nohup blastx -query ../gene_data/53vsX-upgenes.fasta -db /B313/public_db/blast/fish_prot -outfmt 5 -num_threads 8 -out 53vsXup-blastx >ulog_53.out 2>&1 &
nohup blastx -query ../gene_data/54vsY-downgenes.fasta -db /B313/public_db/blast/fish_prot -outfmt 5 -num_threads 8 -out 54vsYdown-blastx >dlog_54.out 2>&1 &
nohup blastx -query ../gene_data/54vsY-upgenes.fasta -db /B313/public_db/blast/fish_prot -outfmt 5 -num_threads 8 -out 54vsYup-blastx >ulog_54.out 2>&1 &
参数解释:
|options|explain|
|:—-|:—-|
|-query|序列,需要比对的序列文件,fasta格式|
|-db|database,在上一步中构建时取得名字,常用为nr、nt两个数据库|
|-outfmt|选择比对输出的类型,有0-14可以选,5是xml文件输出,方便blast2go的进行|
|-out|输出文件|
|-num_threads|线程数量,即使用逻辑CPU的数量,可以加快速度|
注:还有其他的一些参数,可以进一步查询blast官网文档
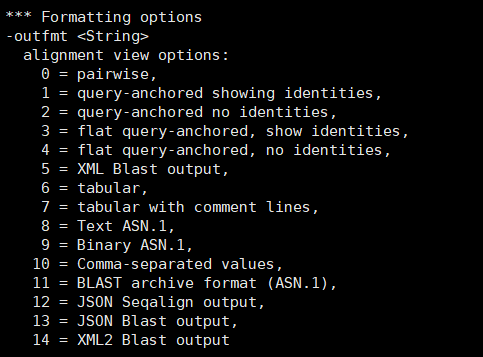
2.4 blast结果
我还没有跑完出来,不太清楚,但是可以先留个位置,等结果出来后进行展示。
总结
- blast基本使用,生信基本工作
- 构建数据库,这一个要掌握好,以后很有可能要自己搭建数据库,满足个性化需求
- 结果文件的输出,选择xml,方便接下来进行注释