1. 前言
在得到了测序结果之后,我们需要评估一下测序的质量,因此我们需要对测序的数据进行统计评价,这里采用的软件组合就是fastqc和multiqc,fastqc用于对每一组的测序结果进行评价并且输出html结果文件,但是当同时有比较多组的测序数据时,一份份html文件去翻阅是非常麻烦的,此时multiqc就派上用场了,multiqc可以对这些结果进行整合,生成一份html文件,方便查阅。
为什么要进行质量评价呢?因为我们后期的所有分析都是基于测序数据展开的,测序数据质量的高低直接影响了我们分析的结果,因此在所有分析步骤之前,我们必须要对测序的数据的质量进行评价,只有达到我们的要求的测序数据才能用于下一步分析。
2. fastqc使用
fastqc是用于统计评估测序数据质量的常用软件,使用命令如下:
fastqc [-o output dir] [--(no)extract] [-f fastq|bam|sam] [-c contaminant file] seqfile1 .. seqfileN
nohup fastqc -o ./fastqc_70/ -f fastq clean_* &
参数解释:
|parameters|explanation|
|:—-|:—-|
|-o|output dir,选择输入结果的文件夹|
|-f|输入文件的格式,支持bam/sam/bam_mapped/sam_mapped/fastq|
|-h|帮助文档,查看参数和使用方式|
还有很多参数未一一列出
结果输出:
输出结果每组都有两个文件,一个是html,一个是zip文件。
3. multiqc使用
命令如下:multiqc /B313/Zjunlin/raw_data/fastqc_70/ -o /B313/Zjunlin/raw_data/multi_qcraw/
参数如下:
结果输出:
4. multiqc结果解读
fastqc的结果解读参数与multiqc一致,所以解读一份就好了。
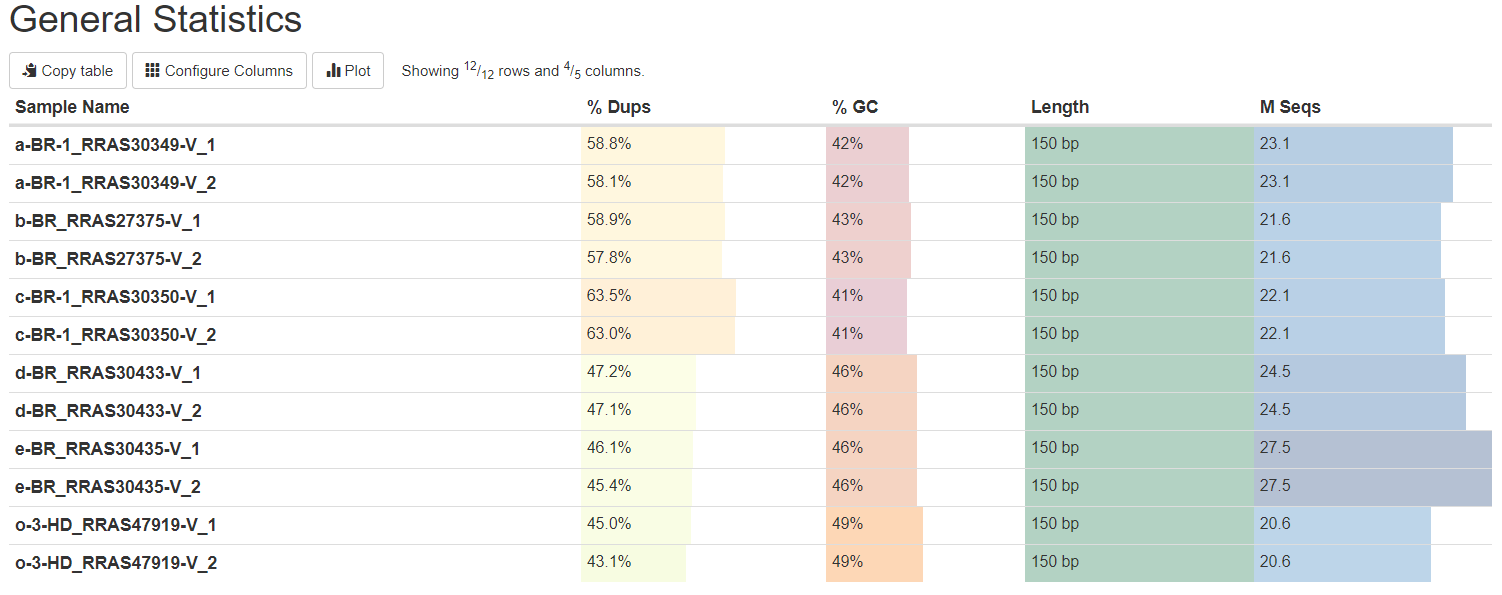
4.1. General Statistics:所有样本数据基本情况统计
%Dups——重复reads的比例
%GC——GC含量占总碱基的比例,比例越小越好
Length——测序长度
M Seqs——总测序量(单位:millions)
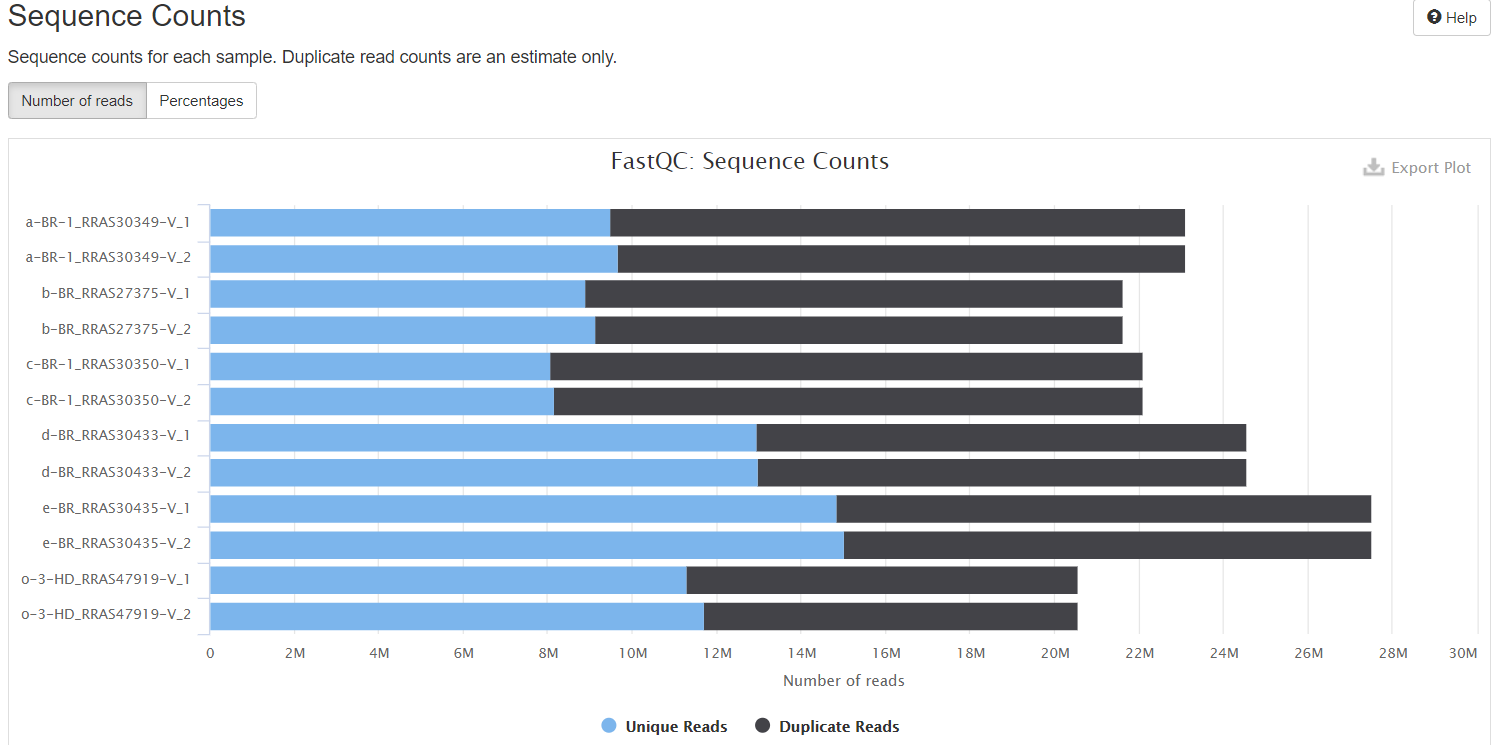
4.2. Sequence Counts:序列数量统计
横坐标——序列的数量
纵坐标——样本
unique reads——蓝色
Duplicate reads——黑色
由图可以看出测序中的一些重复水平,这个重复水平和测序深度以及序列本身的表达情况有关。
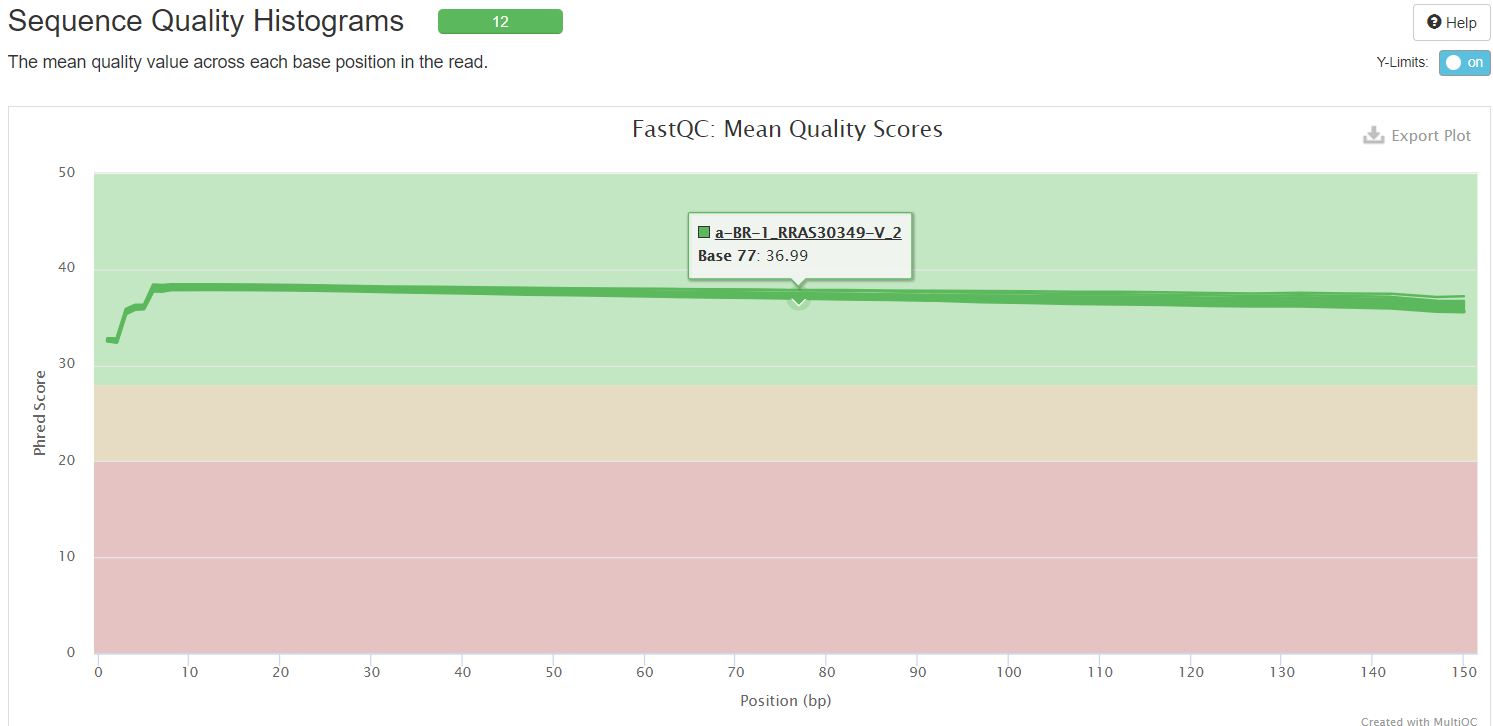
4.3. Sequence Quality Histograms:每个read各位置碱基的平均测序质量
横坐标——碱基的位置
纵坐标——质量分数
质量分数=-10log10p(p代表错误率),所以当质量分数为40的时候,p就是0.0001。此时说明测序质量非常好。
绿色区间——质量很好;
橙色区间——质量合理;
红色区间——质量不好。
可以看到在下面的图中,所有的样本的quality scores都在绿色区间,所以质量很好。
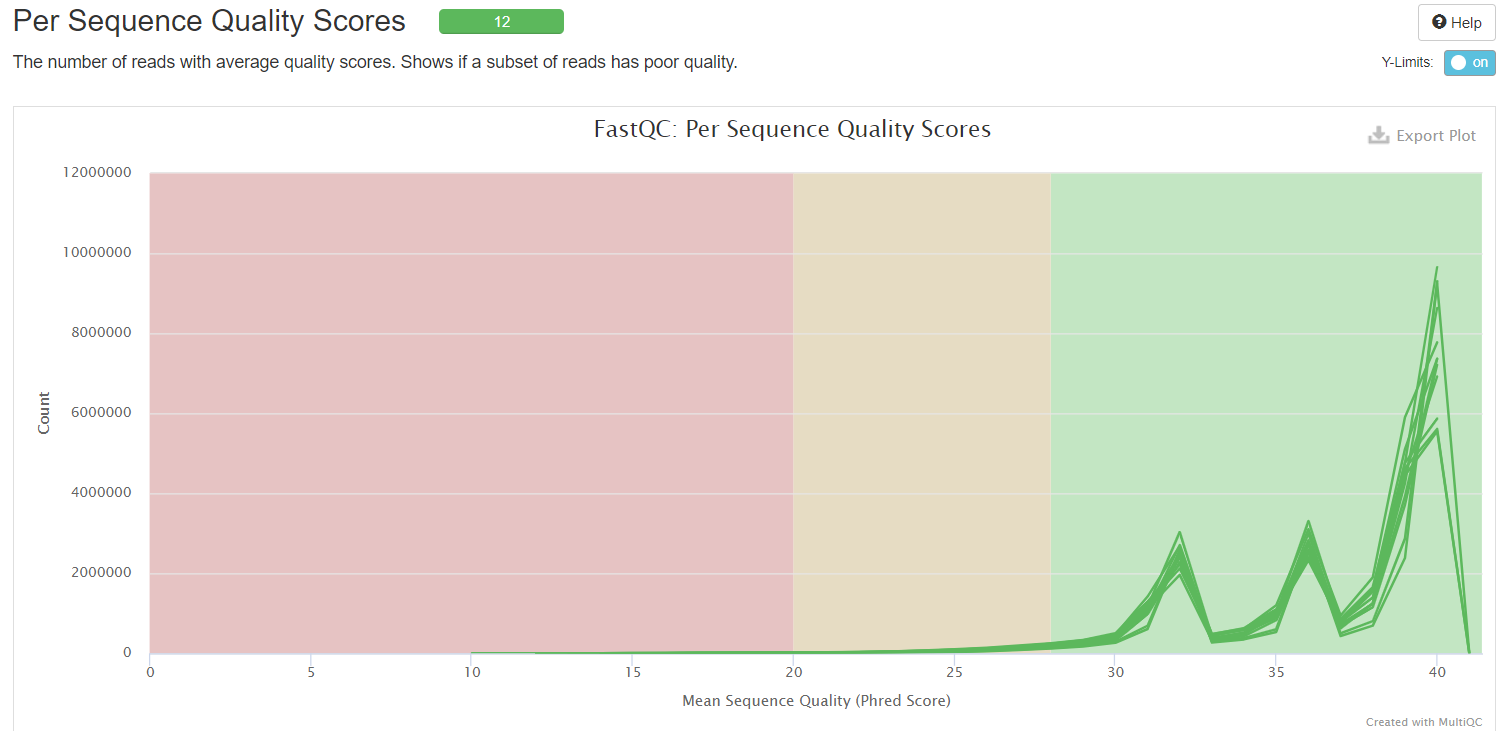
4.4. Per Sequence Quality Scores:具有平均质量分数的reads的数量
横坐标——平均序列质量分数
纵坐标——reads数
绿色区间——质量很好
橙色区间——质量合理
红色区间——质量不好
当峰值小于27时——warning
当峰值小于20时——fail
由此图中可以看出低质量reads占整体reads的比例(估算各颜色区域曲线下面积)。如下图中,几乎所有数值都在绿色区间,所以质量非常好,但是如果存在50%的counts在红色区间,则表示测序结果很差,不可用。
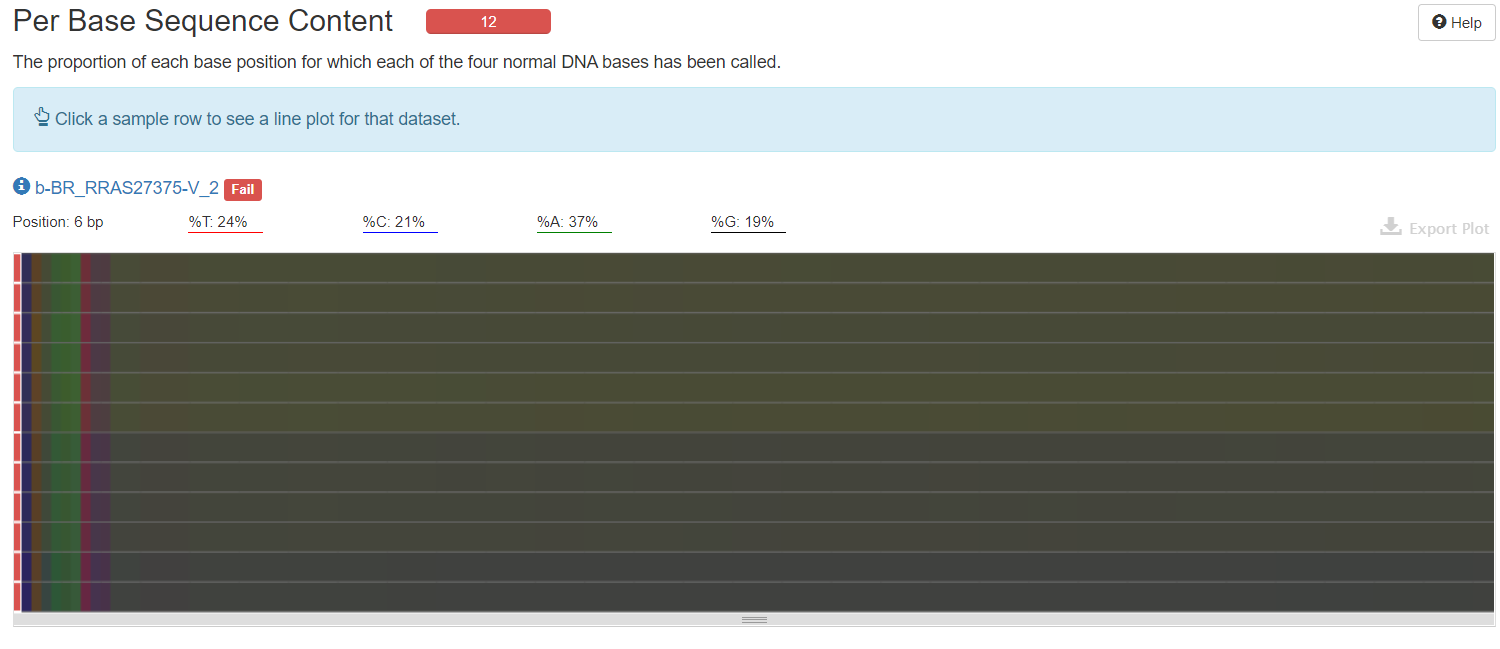
4.5. Per Base Sequence Content:每个read各位置碱基ATCG的比列
对所有reads的每一个位置,统计ATCG四种碱基的分布,
横坐标——碱基位置,
纵坐标——样本。
%A——绿色
%G——紫色
%C——蓝色
%T——红色
如果ATGC在任何位置的差值大于10%——warning
如果ATGC在任何位置的差值大于20%——fail
reads每个位置的颜色显示由4种颜色的比例混合而成,哪一个碱基的比例大,则趋近于这个碱基所代表的颜色。正常情况下每个位置每种碱基出现的概率是相近的。如果ATGC在任何位置的差值大于10%—则warning(橙色),如果ATGC在任何位置的差值大于20%则fail(红色)。在图中可以看出,大概在前9bp的部分,颜色非常不均匀,也就是表示存在AGCT四种碱基的比例差异巨大的地方,这可能有过表达的序列的污染,所以12个样本都是fail。
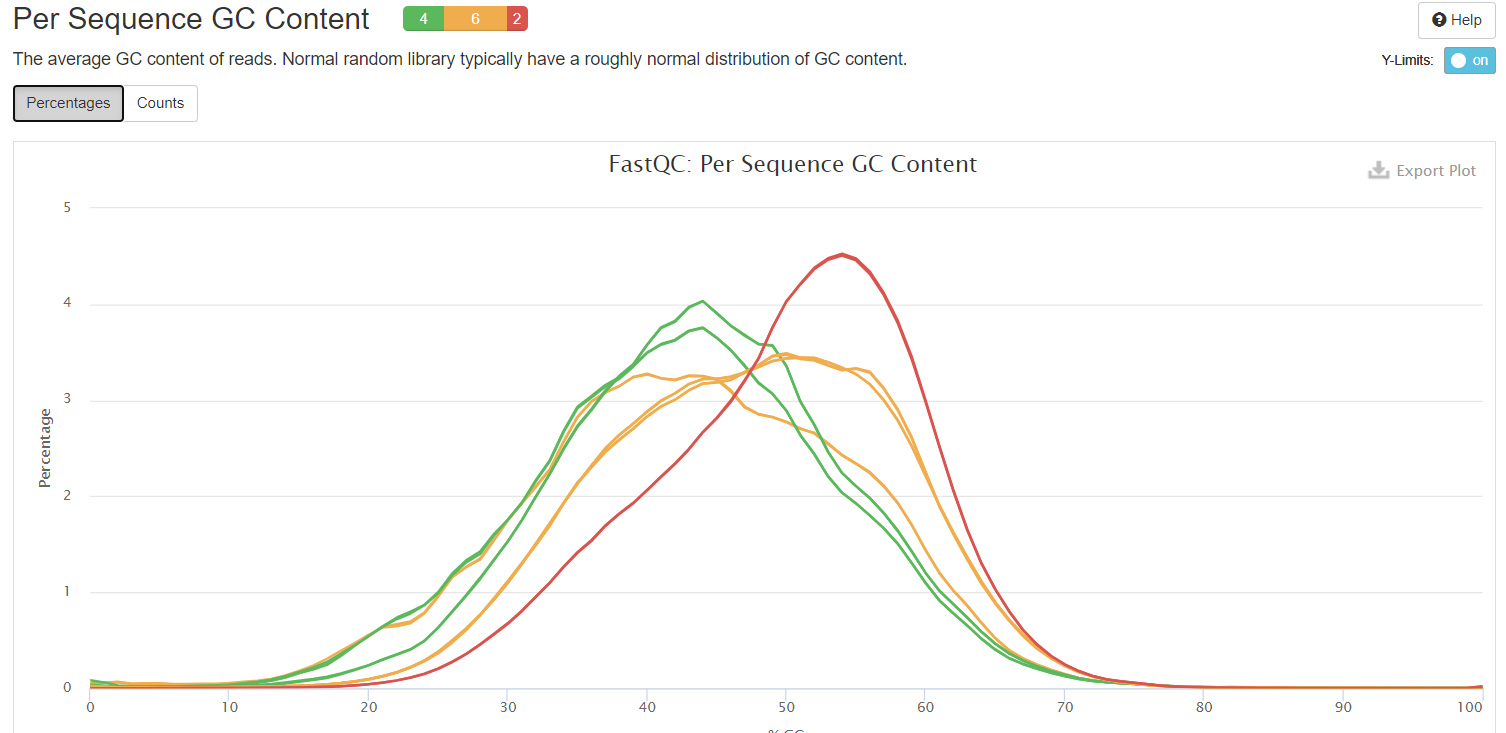
4.6. Per Sequence GC Content:reads的平均GC含量
横坐标——GC含量百分比
纵坐标——数量
正常的样本的GC含量曲线会趋近于正态分布曲线,曲线形状的偏差往往是由于文库的污染或是部分reads构成的子集有偏差(overrepresented reads)。形状接近正态但偏离理论分布的情况提示我们可能有系统偏差。
偏离理论分布的reads超过15%时——warning
偏离理论分布的reads超过30%时——fail
由下图可以看出,有6个样本与理论分布偏离超过了15%,有2个偏离超过了30%,有4个在合理的偏离度内。
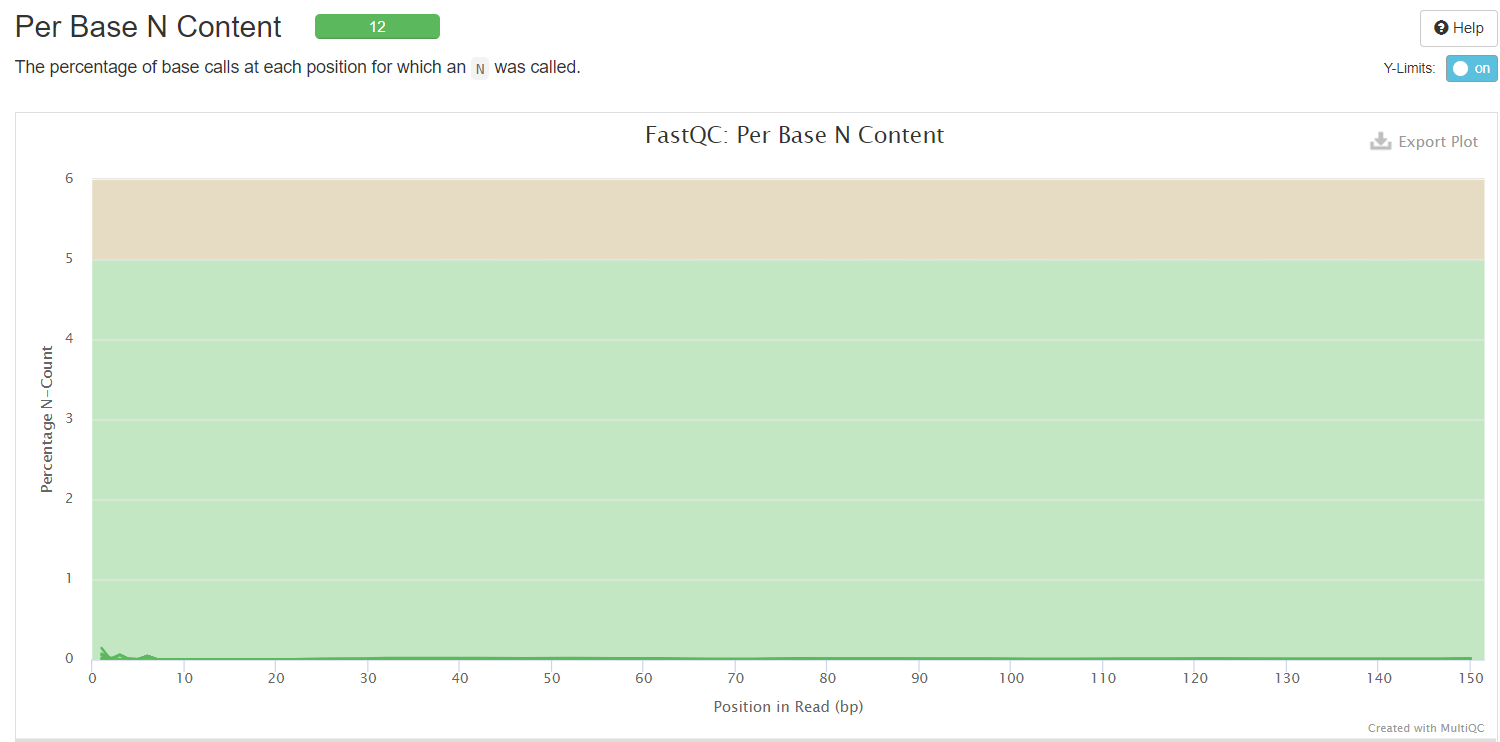
4.7. Per Base N Content:每条reads各位置N碱基含量比例
当测序仪器不能辨别某条reads的某个位置到底是什么碱基时,就会产生“N”,统计N的比率。正常情况下,N值非常小。
横坐标——read中的位置
纵坐标——N的数量比
当任意位置的N的比例超过5%——warning
当任意位置的N的比例超过20%——fail
由图可以看出,所有的样本在所有的位置都是属于绿色区间且数值比例非常小,可以看出测序的质量很高。这应该和现在测序仪工艺的进步,电脑处理能力的提升有着很大的关系,现在的测序仪能够高质量的完成测序。
4.8. Sequence Length Distribution:序列样本长度的分布情况
横坐标——序列长度
纵坐标——数量
reads长度不一致时——warning
reads有0长度时——fail
在这里可以看出测序的读长,也能够分析得出所有的小片段的长度分布情况,依据此我们可以判断测序效果的高低。在这里的结果呈现中,所有的结果都是在150bp,这也是二代测序的特点,测序的读长段,但是片段非常多,也就是通过将基因序列高度片段化分别同时测序,由此提高测序速度。
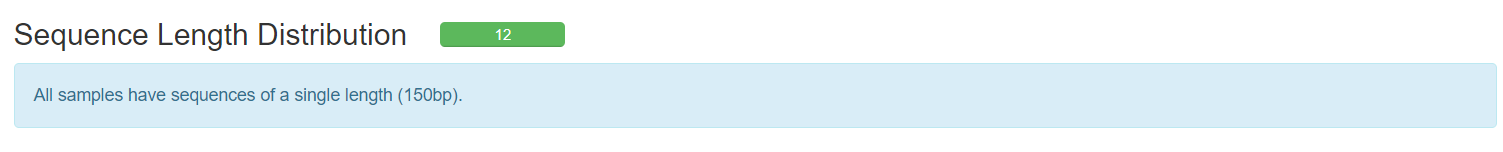
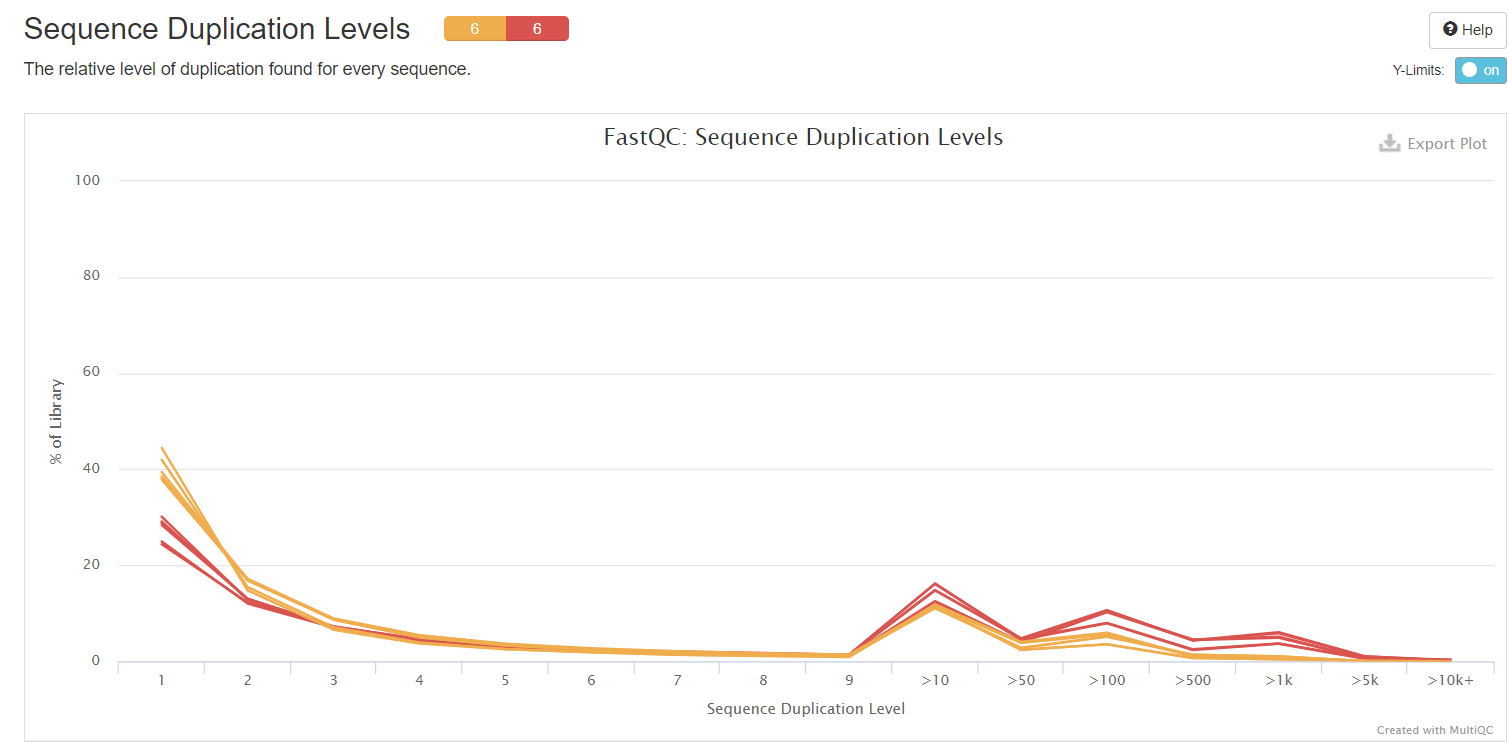
4.9. Sequence Duplication Levels:每个序列的相对重复水平
横坐标——每个序列的相对重复水平
纵坐标——在文库中的比例
当非unique的reads占总数的比例大于20%时——warning
当非unique的reads占总数的比例大于50%时——fail
测序深度越高,越容易产生一定程度的duplication,这是正常的现象,但如果duplication的程度很高,就提示我们可能有bias的存在。
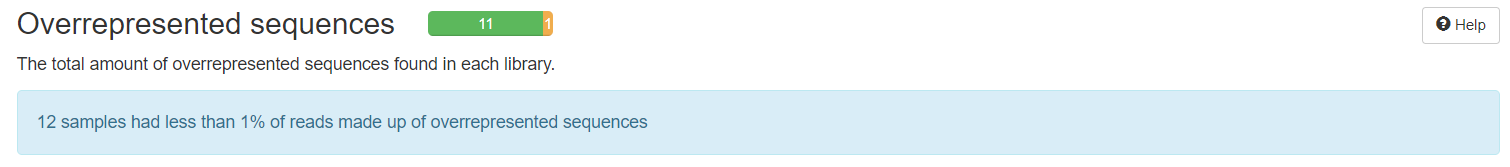
4.10. Overrepresented sequences:文库中过表达序列的比例
横坐标——过表达序列的比例
纵坐标——样本
过表达序列的比例>0.1%——warning
过表达序列的比例>1%——warning
一条序列的重复数,因为一个转录组中有非常多的转录本,一条序列再怎么多也不太会占整个转录组的一小部分(比如1%),如果出现这种情况,不是这种转录本巨量表达,就是样品被污染。这个模块列出来大于全部转录组1%的reads序列,但是因为用的是前100,000条reads,所以其实参考意义不大。
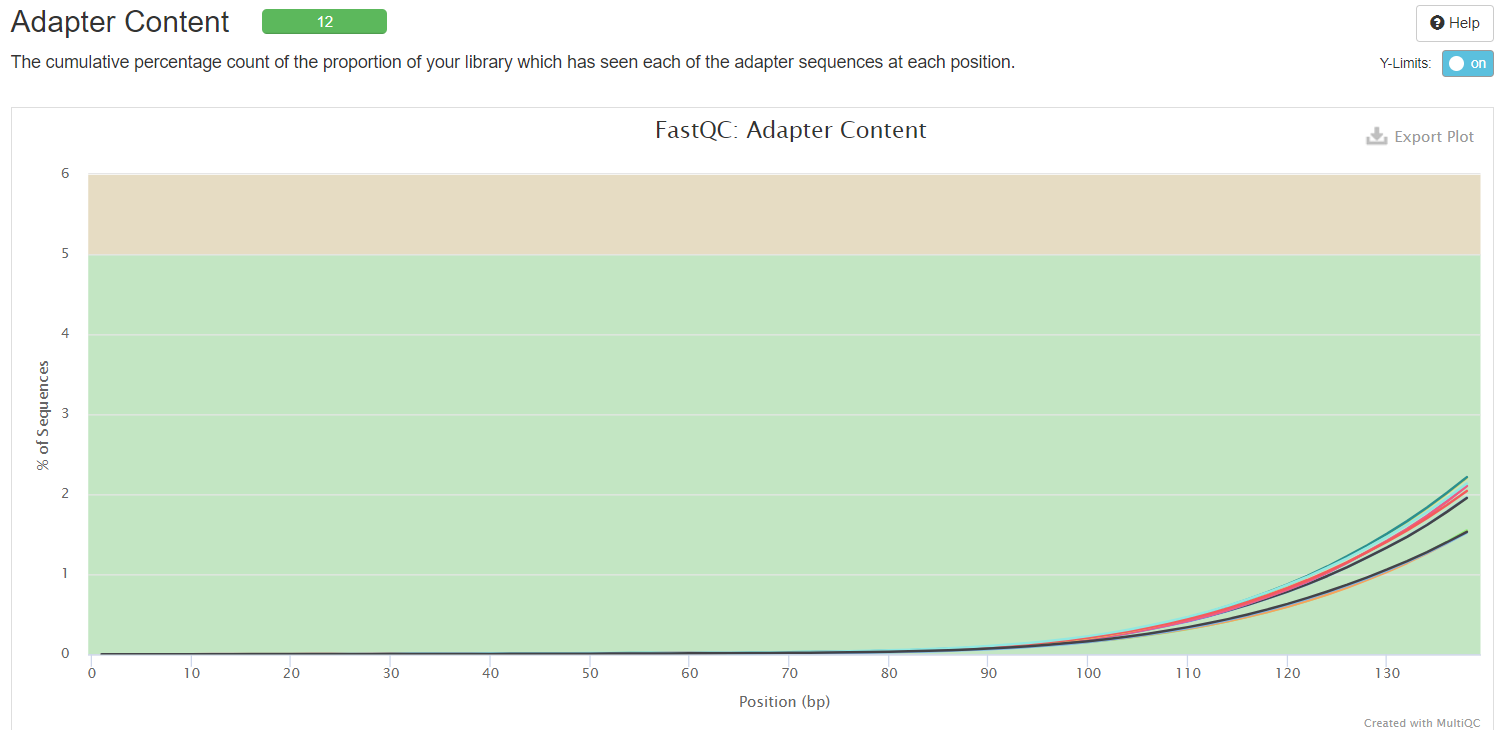
4.11.Adapter Content:接头含量
横坐标——碱基位置
纵坐标——占序列的百分比
百分比>5%——warning
百分比>10%——fail
由图可以看出,所有的样本的接头含量均低于5%,因此结果还算比较良好。