
前言
这是实验室的一个新项目,探讨的是Yellowseabream在生长发育中雄转雌现象的调控基因,这个项目有两个部分,取的样品有脑组织和普通组织,普通组织部分的分析结果已经完成,我负责完成脑组织转录组的分析。因此我将在这篇博客中记录下我在整个项目的过程中的操作,方便以后进行结果整理以及纠错。
1. trimmomatic质控
在拿到测序数据之后,公司的给的data中有raw data也有clean data,但是为了更好的掌控分析,质控还是自己做比较好。这里选择的质控软件是trimmomatic。
commands:
nohup java -jar /B313/public_software/Quality_control/Trimmomatic-0.38/trimmomatic-0.38.jar PE -threads 40 a-BR-1_RRAS30349-V_1.fq a-BR-1_RRAS30349-V_2.fq clean_a-BR-1_RRAS30349-V_1.fq unpaired_a-BR-1_RRAS30349-V_1.fq clean_a-BR-1_RRAS30349-V_2.fq unpaired_a-BR-1_RRAS30349-V_2.fq ILLUMINACLIP:/B313/public_software/Quality_control/Trimmomatic-0.38/adapters/TruSeq3-PE.fa:2:30:10 LEADING:3 TRAILING:3 SLIDINGWINDOW:4:15 MINLEN:50 &
tips:这里指令较长,且需要对每一组进行质控,每次重复输入非常麻烦,因此在第一次输完命令之后可以把命令整理到一个bash脚本中,下次需要重跑时只需要运行bash脚本即可,修改参数可以用Linux文件处理软件sub进行替换修改。
结果文件列表:
输出结果有两种类型文件,一种是clean文件,另一种是unpaired文件。其中clean文件就是质控之后的data,用于下一步分析,unpaired文件是被清理出来的data,只是起一个记录作用。
1.2 fastqc质控可视化
在做质控之前和做完质控之后,要进行数据质量的可视化,用于直观判断质控的效果好坏以及测序数据本身质量的高低。这里采用的是fastqc进行这步工作。
fastqc [-o output dir] [--(no)extract] [-f fastq|bam|sam] [-c contaminant file] seqfile1 .. seqfileN
commands:
nohup fastqc -o ./fastqc_70/ -f fastq clean_* &
output file:
这里输出结果有html和zip文件,只需要查看html文件就可以知道data的质量的统计结果了,但是这里文件众多,一个个查看很麻烦,且不方便进行横向比较,因此可以采用multiqc进行fastqc的结果整合,方便查看。
1.3 multiqc可视化结果整合
commands:
multiqc /B313/Zjunlin/raw_data/fastqc_70/ -o /B313/Zjunlin/raw_data/multi_qcraw/
output file:
这里输出结果是一个html文件和一个data的文件夹,文件夹里面有相关的数据,如果需要自己重新作图的话可以利用这些数据作图。要查看整合的结果只需要把html下载到本地电脑中用浏览器打开查看即可,图片结果的解读在另一篇博客fastqc_multiqc结果解读,可以自行查看
2. Trinity拼接(Trinity-v2.8.4)
Trinity是一款非常成熟的拼接软件,非常受大家欢迎,Trinity的使用方法查看无参转录组分析。
Trinity --seqType fq --left reads_1.fq --right reads_2.fq --CPU 6 --max_memory 20G
commands:
nohup Trinity –seqType fq –max_memory 100G –min_glue 10 –full_cleanup –output trinity_out_dir –left clean_a-BR-1_RRAS30349-V_1.fq clean_b-BR_RRAS27375-V_1.fq clean_c-BR-1_RRAS30350-V_1.fq clean_d-BR_RRAS30433-V_1.fq clean_e-BR_RRAS30435-V_1.fq clean_o-3-HD_RRAS47919-V_1.fq –right clean_a-BR-1_RRAS30349-V_2.fq clean_b-BR_RRAS27375-V_2.fq clean_c-BR-1_RRAS30350-V_2.fq clean_d-BR_RRAS30433-V_2.fq clean_e-BR_RRAS30435-V_2.fq clean_o-3-HD_RRAS47919-V_2.fq –CPU 30 –min_kmer_cov 2 &
这里需要等待比较长的计算时间,并且在结果出来之后要进行统计,要是统计结果不太好则要重新调整参数重跑。所以还是把质量写进脚本run_trinity.sh比较方便。
output files: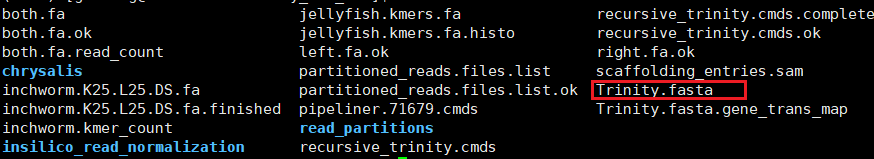
输出的结果包括了三部分,分别是Trinity当中三部分程序的独立结果。在所有结果中最为重要的是Trinity.fasta文件,这就是我们要的最终的拼接结果的文件,后续分析以此为基础,我们需要先评估拼接的质量高低,满足要求就开始下一步定量分析。
2.1 TrinityStats.pl结果统计
/B313/public_software/trinityrnaseq-Trinity-v2.8.4/util/TrinityStats.pl <result_fasta_file>
commands:
/B313/public_software/trinityrnaseq-Trinity-v2.8.4/util/TrinityStats.pl ./Trinity.fasta
output: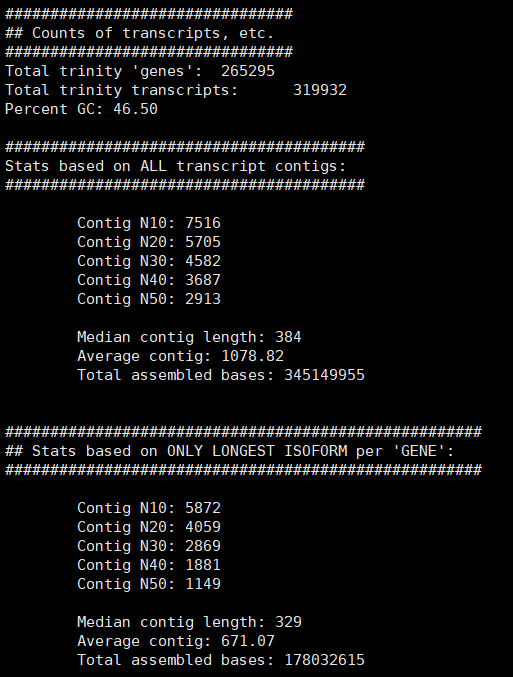
| total genes | total transcripts | Contig N50 | Percent GC |
|---|---|---|---|
| 265295 | 319932 | 2913 | 46.50 |
由上结果来看,拼接出来的转录本数量有点多,这样对于在时候做差异分析时不利,下一回可以考虑在指控的时候把minlen参数设高一些,这样子对于后期的分析比较有利。这里拼接的N50是2913,还不错。
2.2 cd-hit去冗余
commands:
cd-hit-est -i input.fasta -o output.fasta -c 0.90 -n 8 -T 40
output: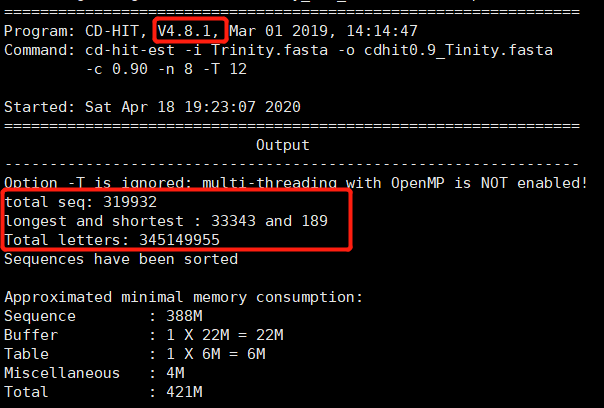
2.3 corset聚类去冗余
commands:
corset [options] bowtie.bam
- 注意使用python2.7环境
3. RSEM_bowtie2定量分析与mapping
commands:
1 | nohup /B313/public_software/trinityrnaseq-Trinity-v2.8.4/util/align_and_estimate_abundance.pl --transcripts cdhit_new_Trinity.fasta --seqType fq --est_method RSEM --aln_method bowtie --trinity_mode --prep_reference --output_dir rsem_outdir_a --left clean_a-BR-1_RRAS30349-V_1.fq --right clean_a-BR-1_RRAS30349-V_2.fq >nohup_a.out 2>&1 & |
tips: Each group needs to align independently.
results:
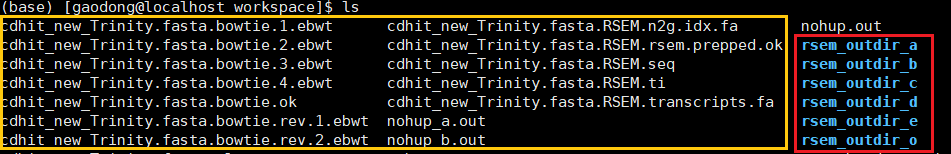
黄色框中是RSEM定量的一些中间文件,红色框中是结果输出文件夹

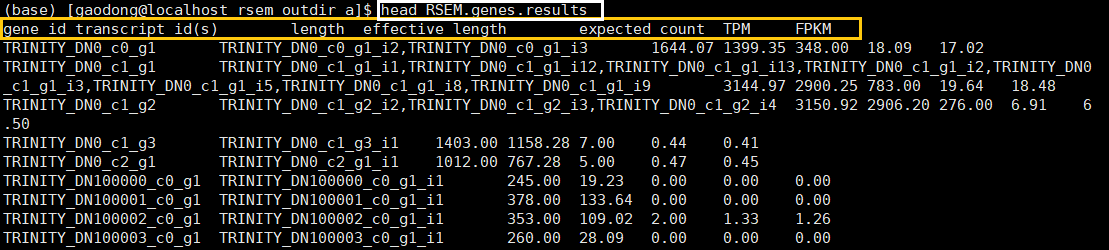
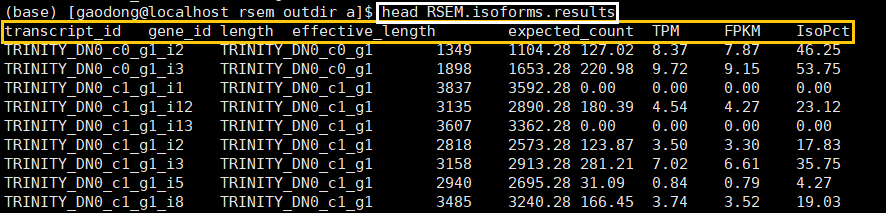
genes.results和isoforms.results分别是基于基因和转录本水平的定量结果,这两者的区别可以在下面两幅图中看出来。isoforms.results中包含了转录本ID,基因ID,转录本长度,有效长度,expected_count,TPM,FPKM和IsoPct(该转录本表达量占基因总表达量的百分比)。genes.results中的内容与之类似,只是少了IsoPct。
接下来我们可以将FPKM提取出来,然后进行聚类分析,查看一下现有的各组样品之间的一个相关性,这类可以采用clustermap来展示。